
Il Regolamento UE n. 2017/745 (MDR) sui dispositivi medici: ambito di applicazione e periodo transitorio. Dispositivi medici e AI.
Il Regolamento UE n. 2017/745 sui dispositivi medici (Medical Device Regulation, “MDR”) che ha abrogato le precedenti direttive europee in materia e i Decreti legislativi n. 507/92 e n. 46/97, ha introdotto importanti modifiche alla disciplina della produzione e commercializzazione dei dispositivi medici. L’entrata in vigore del MDR è stata posticipata dapprima dal Regolamento UE 2020/561 emesso a seguito dell’epidemia Covid, e successivamente dal Regolamento (UE) 2023/607. che ha esteso il periodo di transizione in modo da concedere agli operatori più tempo per condurre le necessarie procedure di valutazione della conformità per i dispositivi legacy, cioè i dispositivi immessi sul mercato dopo il 26 maggio 2021 (data di entrata in vigore del MDR) e certificati ai sensi delle precedenti direttive. L’MDR ha ampliato notevolmente la definizione di dispositivo medico, allargando quindi il novero dei prodotti a cui si applica la nuove normativa. Da ultimo, il Regolamento Ue 2024/1689 sull’Intelligenza Artificiale (“AI Act”) si applica anche ai DM software, prevedendo una serie di obblighi stingenti per gli operatori, in particolare per i sistemi definibili ad alto rischio. Analizziamo l’ambito di applicabilità del MDR, il periodo transitorio e l’applicazione dell’AI ACT ai DM.
1. Il Regolamento UE n. 2017/745 sui dispositivi medici (MDR)
Il settore dei dispositivi medici (DM) riveste in ambito europeo una grande importanza nell’assistenza sanitaria, contribuendo in modo rilevante al miglioramento del livello di protezione della salute attraverso lo sviluppo di soluzioni innovative per la diagnosi, la prevenzione, le cure e la riabilitazione.
Il 26 maggio 2021 (a seguito del posticipo di un anno dovuto all’emergenza sanitaria Covid-19) è entrato definitivamente in vigore il nuovo Regolamento UE n. 2017/745 sui dispositivi medici (Medical Device Regulation, “MDR”) che ha abrogato la precedente direttiva 93/42/CE (di seguito la “Direttiva“) e conseguentemente i Decreti legislativi n. 507/92 e n. 46/97.
L’MDR ha introdotto importanti modifiche alla disciplina della produzione e commercializzazione dei DM, aumentando il livello di sicurezza e prestazione degli stessi e ridisegnando i compiti e le responsabilità degli operatori economici coinvolti nell’intera filiera, chiamati oggi a svolgere un ruolo molto più proattivo nel corso dell’intera vita del dispositivo.
In questo senso, l’MDR ha modificato profondamente la normativa preesistente su diversi aspetti, quali l’immissione sul mercato, la supervisione degli organismi notificati, le procedure di valutazione della conformità dei prodotti, le indagini e le valutazioni cliniche, la valutazione del rischio, nonché la sorveglianza del mercato.
La necessità di modificare la normativa di settore nasceva dall’esigenza di assicurare, anche mediante una sempre maggiore armonizzazione, il buon funzionamento del mercato interno all’Unione Europea, innalzando al contempo gli standard di qualità e sicurezza dei DM. Ciò nel contesto di un quadro legislativo che pone l’UE quale garante della salute pubblica e della salute e sicurezza dei pazienti.
L’MDR prevede un elenco dettagliato dei requisiti di sicurezza e prestazione che tutti i dispositivi medici immessi in commercio in Europa devono rispettare al fine di garantire un elevato livello di qualità, ed aumenta le responsabilità degli operatori coinvolti nella catena di fornitura, intensificando in particolare gli obblighi di controllo soprattutto sulle prestazioni del DM lungo tutto il suo ciclo di vita.
Gli utilizzatori, siano essi strutture sanitarie o operatori economici, potranno pertanto utilizzare dispositivi il cui livello di sicurezza e prestazione è maggiormente validato, ma le cui istruzioni d’uso da rispettare saranno molto più dettagliate e precise. Ciò anche alla luce del fatto che l’MDR ha stabilito nuove regole di classificazione dei DM.
L’MDR è direttamente applicabile negli Stati membri UE, i quali sono tenuti ad armonizzare la disciplina nazionale sui DM. In Italia, la L. n. 53/2021 ha stabilito i principi e i criteri direttivi che devono essere rispettati per adeguare la normativa nazionale al nuovo quadro legislativo introdotto dall’MDR. Tale adeguamento è quindi avvenuto con il D.lgs. n. 137 del 5 agosto 2022.
2. L’entrata in vigore del MDR
2.1 La disciplina originaria e il Regolamento UE 2020/561
L’MDR è stato pubblicato nella Gazzetta ufficiale dell’Unione europea il 5 maggio 2017 ed è entrato in vigore il 26 maggio 2017. Si è peraltro posto il problema di disciplinare i DM già immessi legittimamente sul mercato in precedenza, nel vigore della Direttiva (c.d. dispositivi legacy).
Per “dispositivi legacy” si intendono:
- I DM rientranti nella classe I ai sensi della Direttiva, per i quali è stata redatta una dichiarazione di conformità CE prima del 26 maggio 2021 e per i quali la procedura di valutazione della conformità ai sensi dell’MDR richiede il coinvolgimento di un ON;
- I DM coperti da un certificato CE valido rilasciato ai sensi della Direttiva prima del 26 maggio 2021.
E’ opportuno soffermarsi sui termini di “immissione in commercio” e “messa in servizio”, data la loro diretta attinenza all’entrata in vigore del MDR.
Secondo il MDR, per “immissione in commercio” di un dispositivo si intende la prima messa a disposizione di un dispositivo sul mercato dell’Unione, ovvero la prima fornitura di un dispositivo, diverso da un dispositivo oggetto di indagine, per la distribuzione, il consumo o l’uso sul mercato dell’Unione nel corso di un’attività commerciale, a titolo oneroso o gratuito (art. 2 MDR). La data di fabbricazione di un DM medico non è quindi un requisito sufficiente per determinare il momento della sua immissione in commercio.
In proposito, la “Guida Blu” del 2016 ha chiarito quanto segue:
- un prodotto si considera immesso in commercio quando è messo a disposizione per la prima volta sul mercato dell’Unione; ciò avviene non nel momento in cui si immette fisicamente sul mercato il prodotto, ma quando si conclude (verbalmente o per iscritto) un accordo commerciale tra le parti per il trasferimento della proprietà o del possesso del prodotto, a titolo oneroso o gratuito, indipendentemente dalla consegna materiale del prodotto;
- tale operazione è riservata al fabbricante o all’importatore (i quali sono dunque gli unici operatori economici a che possono immettere prodotti sul mercato);
- la prima messa a disposizione è riferita al singolo pezzo (a prescindere dal fatto che sia un esemplare unico o in serie) e non alla categoria di prodotti.
Pertanto, quando un fabbricante o un importatore fornisce un prodotto a un distributore o a un utilizzatore finale per la prima volta, tale operazione è sempre designata in termini giuridici come “immissione sul mercato”.
Qualsiasi operazione successiva, ad esempio da distributore a distributore o da distributore a utilizzatore finale, è definita invece “messa a disposizione”. Anche la prima messa a disposizione è riferita al singolo pezzo e non alla categoria di prodotti, a prescindere dal fatto che si tratti di un esemplare unico o in serie.
Non si ha immissione di un DM sul mercato nel sito in cui un prodotto sia presente nei magazzini del fabbricante (o del suo rappresentante autorizzato stabilito nell’Unione) o dell’importatore, ma non venga ancora messo a disposizione; ciò in quanto l’arrivo del DM al magazzino dell’importatore è un fatto fisico, non sufficiente di per sé a configurare l’immissione sul mercato, che avviene invece solo nel momento in cui l’importatore stipula con altro soggetto un contratto di trasferimento della proprietà o del possesso.
Secondo la Guida Blu non si considerano come immissione sul mercato i casi in cui un prodotto sia:
- fabbricato per uso personale;
- acquistato da un consumatore in un paese terzo mentre era fisicamente presente in questo paese e portato nell’UE da tale consumatore per suo uso personale:
- trasferito dal fabbricante in un paese terzo a un mandatario nell’Unione che il fabbricante ha designato per garantire che il prodotto sia conforme alla normativa di armonizzazione dell’Unione;
- introdotto da un paese terzo nel territorio doganale dell’UE, in transito, posto in zona franca, in deposito, in custodia temporanea o vincolato ad altro regime doganale speciale (ammissione temporanea o perfezionamento attivo):
- fabbricato in uno Stato membro per l’esportazione in un paese terzo (compresi componenti forniti a un fabbricante per l’incorporazione in un prodotto finale da esportare in un paese terzo);
- trasferito per la prova o la convalida di unità preproduzione considerate ancora in fase di fabbricazione;
- esposto o utilizzato in condizioni controllate in occasione di fiere, mostre o dimostrazioni;
- presente nei magazzini del fabbricante (o del suo rappresentante autorizzato stabilito nell’Unione) o dell’importatore, ma non ancora messo a disposizione, e quindi non fornito per la distribuzione, il consumo o l’uso, salvo diversa disposizione della vigente normativa di armonizzazione dell’Unione.
La Guida Blu ha altresì precisato che I prodotti messi in vendita da operatori online stabiliti nell’Ue sono considerati immessi sul mercato dell’Unione, indipendentemente dal soggetto che li ha immessi sul mercato, mentre i prodotti messi in vendita online da venditori stabiliti al di fuori dell’Ue sono considerati immessi sul mercato dell’Unione se le vendite sono specificamente destinate a consumatori o altri utilizzatori finali dell’Ue. Sebbene la Guida Blu non specifichi cosa si debba intendere per “messa in vendita”, si può ritenere che tale situazione si concretizzi quando le informazioni che appaiono sul sito siano sufficientemente dettagliate (tipologia di dispositivo, lotto, prezzo, modalità di consegna etc.) da poter configurare un contenuto minimo contrattuale.
Ciò premesso, il MDR prevedeva originariamente un periodo di transizione che si sarebbe dovuto concludere il 26 maggio 2020, data prevista di applicazione del MDR. Intervenuta nel frattempo la crisi sanitaria causata dall’epidemia dovuta al COVID-19, tale data è stata in un primo tempo posticipata di un anno (ovvero al 26 maggio 2021) dal Regolamento UE 2020/561, entrato in vigore il 24 aprile 2020.
L’epidemia ha infatti richiesto un aumento della disponibilità di DM di vitale importanza e di conseguenza è stato ritenuto fondamentale assicurare il corretto funzionamento del mercato dell’Unione, evitando quelle perturbazioni che si sarebbero potute generare dall’impossibilità, in tale contesto, di garantire l’attuazione e la corretta applicazione del MDR.
Pertanto, l’art. 120 del MDR , modificato dal Regolamento UE 2020/561, stabiliva che:
- i DM di classe I ai sensi della Direttiva, che dovevano essere classificati in una classe superiore secondo il MDR (dovendo quindi coinvolgere un Organismo Notificato (ON) nella valutazione della conformità) potevano essere immessi in commercio (ovvero, ai sensi dell’art. 2, punto 28 MDR, messi a disposizione sul mercato dell’Unione) conformemente alla Direttiva, fino al 26 maggio 2024, a condizione che la dichiarazione di conformità fosse stata redatta dal fabbricante prima del 26 maggio 2021;
- i DM di classe superiore alla I (IIa, IIb e III) potevano essere immessi in commercio conformemente alla Direttiva fino alla scadenza del Certificato CE emesso dall’ON (al più tardi fino al 26 maggio 2024).
In entrambi i casi, per poter beneficiare di questo periodo transitorio, occorreva rispettare le seguenti condizioni (che devono essere rispettate anche a seguito della successiva modifica del periodo transitorio (v. par 2.2)).
- non dovevano essere apportati ai DM “cambiamenti significativi” (v. par. 2.3.1) né deve essere cambiata la destinazione d’uso;
- dovevano comunque essere rispettate le prescrizioni del MDR in materia di sorveglianza post-commercializzazione, vigilanza, sorveglianza del mercato e registrazione degli operatori economici sulla piattaforma EUDAMED.
I DM che beneficiavano del periodo transitorio potevano essere messi in servizio (ovvero, ai sensi dell’art. 2, punto 29 MDR, resi disponibili all’utilizzatore finale in quanto pronti per il primo utilizzo sul mercato dell’Unione secondo la destinazione d’uso) fino al 27 maggio 2025 (c.d. sell-off date); a partire da tale data, tutti i DM avrebbero dovuto essere conformi al MDR.
Tuttavia, data l’impossibilità da parte degli ON di garantire la valutazione della conformità all’MDR della grande quantità di dispositivi nei tempi previsti, e dal momento che molti fabbricanti non erano ancora pronti alla transizione, vi era il forte rischio che molti DM, già coperti da certificato secondo la Direttiva, non avrebbero ottenuto la certificazione conforme ai sensi del MDR entro la fine del periodo transitorio, il che avrebbe determinato una penuria di DM a disposizione sul mercato dell’unione.
2.2 La disciplina del periodo transitorio prevista dal Regolamento (UE) 2023/607
Il 20 marzo 2023 il Parlamento Europeo ha quindi pubblicato il Regolamento (UE) 2023/607, che ha ulteriormente modificato l’art. 120 par. 3 MDR, estendendo nuovamente il periodo di transizione, in modo da concedere ai fabbricanti e agli ON più tempo per condurre le necessarie procedure di valutazione della conformità per i dispositivi legacy “coperti” da un certificato o da una dichiarazione di conformità rilasciata ai sensi della Direttiva.
Il nuovo Regolamento ha previsto che i dispositivi legacy con certificato CE valido secondo la Direttiva rilasciato prima del 26 maggio 2021 e i dispositivi di classe I secondo la Direttiva, con dichiarazione di conformità CE emessa prima del 26 maggio 2021, che con il MDR passano ad una classe superiore (che richiede l’intervento di un ON nella valutazione di conformità), possono immettere tali DM sul mercato (v. par. 2.3) anche dopo tale data, beneficiando di un periodo transitorio prolungato.
L’estensione del periodo in cui i dispositivi legacy possono essere legittimamente immessi sul mercato o messi in servizio è collegata alla classe di rischio del DM, determinata ai sensi delle regole del MDR (e non della previgente Direttiva). In particolare:
- i DM legacy di classe III o di classe IIb impiantabili possono essere immessi sul mercato o messi in servizio fino al 31 dicembre 2027 (ad eccezione di materiali per sutura, graffette, materiali per otturazioni dentarie, apparecchi ortodontici, corone dentali, viti, cunei, placche e protesi, fili, chiodi, clip e connettori);
- i DM legacy di classe IIb (non impiantabili), classe IIa, classe I sterili o con funzione di misura(secondo le regole dell’allegato VIII dell’MDR) possono essere immessi sul mercato o messi in servizio fino al 31 dicembre 2028;
- i dispositivi per i quali la procedura di valutazione della conformità secondo la Direttiva non richiedeva l’intervento di un ON e per i quali la procedura di valutazione della conformità a norma del MDR richiede l’intervento di un ON, possono essere immessi sul mercato o messi in servizio fino al 31 dicembre 2028.
- i dispositivi impiantabili su misura di classe III possono essere immessi sul mercato o messi in servizio fino al 26 maggio 2026.
Per beneficiare dell’estensione prevista, l’art. 120 commi 3 e 3-quarter MDR, così come modificati dal Regolamento 2023/607, prevedono debbano essere soddisfatte le seguenti condizioni:
a) i DM devono continuare ad essere conformi alla Direttiva;
i DM non devono presentare cambiamenti significativi nella progettazione e nella destinazione d’uso;
c) i DM non devono presentare un rischio inaccettabile per la salute o la sicurezza di pazienti, utilizzatori o altre persone o per altri aspetti della protezione della salute pubblica;
d) entro il 26 maggio 2024 il fabbricante deve avere istituito un sistema di gestione nella qualità conformemente all’art. 10, par. 9 MDR;
e) entro il 26 maggio 2024 il fabbricante o il mandatario deve avere presentato una domanda formale a un ON conformemente all’allegato VII, punto 4.3, primo comma MDR, per la valutazione della conformità del dispositivo (o di un dispositivo destinato a sostituirlo) ed entro il 26 settembre 2024 l’ON e il fabbricante devono avere firmato un accordo scritto conformemente all’allegato VII, punto 4.3, secondo comma MDR.
Tali aspetti saranno approfonditi nel par. 2.3.
Le disposizioni del Regolamento (UE) 2023/607 hanno lo scopo di facilitare il permanere in commercio del DM legacy, evitando possibili interruzioni nella loro commercializzazione a causa di eventuali ritardi nel processo di valutazione da parte degli ON, ed il conseguente scadimento della garanzia della tutela della salute dei pazienti che ne beneficiano (oltre che il conseguimento disagio ed utilizzatori e fabbricanti).
Un dispositivo legacy coperto da un certificato con scadenza dopo il 20 marzo 2023 e prima del 26 maggio 2024, può beneficiare della proroga fino al 26 maggio 2024 se il fabbricante non ha intenzione di procedere con la certificazione MDR. Pertanto, la validità dei Certificati relativa ai DM legacy per cui non viene presentata domanda ai sensi del MDR decade il 26 maggio 2024.
Dal 26 maggio 2024, i DM legacy per i quali non è stata presentata una domanda di registrazione secondo il MDR, non possono più essere immessi sul mercato dell’UE. Per tali DM, l’ON rilascia una comunicazione di “fine periodo transitorio”, che servirà a informare i fabbricanti e i distributori che il DM non può più essere commercializzato a partire dalla data indicata.
In ogni caso, non devono essere intervenuti cambiamenti significativi nella progettazione e nella destinazione d’uso del DM, e non deve essersi verificato un rischio inaccettabile per la salute o sicurezza (v. par. 2.1 ). L’Organismo che ha emesso il certificato secondo la Direttiva può confermare per iscritto, dopo aver esaminato la descrizione del fabbricante della modifica (proposta), che l’implementazione della modifica non rappresenta un cambiamento significativo nel design o nell’uso previsto ai sensi del MDR e che il certificato rimane valido fino alla fine del periodo di transizione. Tale conferma corregge o integra le informazioni sul certificato esistente, ma non lo modifica né lo sostituisce.
Qualora invece il fabbricante intenda immettere sul mercato un DM che presenti cambiamenti significativi (v. par. 2.3.1) rispetto al DM precedente (“device intended to substitute that device”), il periodo transitorio è applicabile solo al dispositivo preesistente. Il nuovo DM deve essere sottoposto a valutazione completa di conformità secondo l’MDR prima di essere immesso sul mercato.
Successivamente alla certificazione MDR del dispositivo sostitutivo, il DM preesistente e quello sostitutivo potranno essere immessi sul mercato in parallelo fino alla fine del relativo periodo transitorio. Quando il fabbricante presenta la domanda per un DM destinato a sostituire un DM precedente, deve identificare il DM sostitutivo e quello precedente che si intende sostituire; la documentazione tecnica del DM sostitutivo può essere presentata in una fase successiva.
Il Regolamento (UE) 2023/607 ha inoltre eliminato la sell-off date del 27 maggio 2025, consentendo quindi ai distributori di mettere a disposizione sul mercato o mettere in servizio i dispositivi legacy già immessi legittimamente sul mercato anche successivamente a tale data.
Pertanto, i dispositivi immessi legittimamente sul mercato secondo la Direttiva prima del 26 maggio 2021 e quelli immessi legittimamente sul mercato dopo tale data durante il periodo transitorio previsto dall’art. 120 MDR (ovvero a seconda dei casi fino al 31 dicembre 2027 o fino al 31 dicembre 2028) possono continuare a essere messi a disposizione sul mercato o a essere messi in servizio senza limitazione di tempo (entro il limite di durata di conservazione o di scadenza del dispositivo).
2.3 Gli obblighi dei fabbricanti di dispositivi legacy nel periodo transitorio
Vediamo gli obblighi in capo ai fabbricanti di DM legacy durante il periodo transitorio, alla luce del Regolamento (UE) 2023/607 e delle linee guida Mdcg 2021-25.
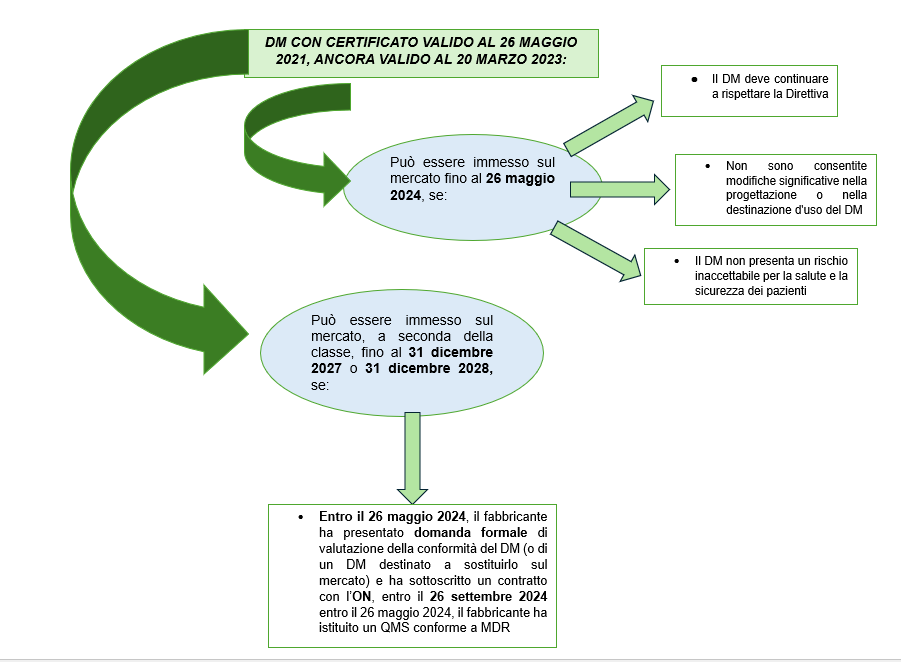
Se un DM, alla data del 26 maggio 2021, aveva un certificato CE valido secondo la Direttiva, e ancora valido alla data di pubblicazione del Regolamento (UE) 2023/607 (20 marzo 2023), tale certificato resta valido fino al 26 maggio 2024, se il dispositivo:
- rispetta la Direttiva
- non presenta modifiche significative nella progettazione o nella destinazione d’uso;
- non presenta un rischio inaccettabile per la salute e la sicurezza dei pazienti;
- viene adeguatamente sorvegliato.
Al fabbricante viene data la possibilità di immettere sul mercato il dispositivo anche dopo tale data, ovvero:
- fino al 31 dicembre 2027 per tutti i dispositivi di classe III e per i dispositivi impiantabili di classe IIb;
- fino al 31 dicembre 2028, per i dispositivi della classe IIb diversi da quelli sopra, per i dispositivi della classe IIa e per i dispositivi della classe I sterile o con funzione di misura.
In tale periodo, il dispositivo deve continuare ad essere conforme alla Direttiva, non deve subire cambiamenti progettuali o di scopo significativi (v. par. 2.3.1) e non deve rappresentare un rischio inaccettabile per la salute di utilizzatori e pazienti o per altri aspetti inerenti alla salute pubblica.
Più esattamente, come previsto dalle linee guida Mdcg 2021-25, i fabbricanti DM legacy devono:
- istituire tenere aggiornato un sistema di sorveglianza post-commercializzazione, ai sensi dell’art. 83 MDR;
- definire un Piano di Sorveglianza post commercializzazione ai sensi dell’art. 84 MDR;
- redigere il rapporto sulla sorveglianza post-commercializzazione per i DM legacy di classe I, ai sensi dell’art. 85 MDR;
- redigere il Rapporto periodico di aggiornamento sulla sicurezza per i DM Legacy di classe IIa, IIb e III, ai sensi dell’art. 85 MDR;
- adottare immediatamente le azioni correttive, ai sensi dell’art. 10, par. 12 MDR;
- segnalare gli incidenti gravi e le tendenze ai sensi degli artt. 87 e 88 MDR;
- analizzare gli “incidenti gravi” e le “azioni correttive” ai sensi dell’art. 89 MDR;
- registrare il DM e la propria posizione di fabbricante in Eudamed (quando sarà pienamente operativa).
Sono invece esclusi per i fabbricanti di DM legacy alcuni obblighi previsti dal MDR, quali in particolare:
- la nomina della persona responsabile della normativa ai sensi dell’art. 15 MDR;
- la revisione della documentazione tecnica alla luce degli Allegati II e III MDR (salvo quanto previsto con riguardo all’obbligo di redigere un piano di sorveglianza post-commercializzazione, il rapporto sulla sorveglianza post-commercializzazione e il rapporto periodico di aggiornamento sulla sicurezza);
- il rispetto dei nuovi obblighi documentali e informativi previsti dall’art. 18 MDR a favore del portatore di un dispositivo impiantabile, salvo il rispetto delle eventuali e diverse norme nazionali;
- l’identificazione della catena di fornitura ai sensi dell’articolo 25 MDR, salvo l’obbligo di rispettare ulteriori normative europee e nazionali sulla tracciabilità dei prodotti (si pensi ad es. alla Direttiva 2001/95/CE sulla sicurezza generale dei prodotti);
- l’attribuzione di un Codice Udi ai sensi dell’art. 27 MDR.
La proroga della validità del certificato avviene automaticamente per legge, a condizione che siano soddisfatti i requisiti di cui all’art. 120, comma 3, MDR, ovvero:
a) i DM devono continuare ad essere conformi alla Direttiva;
b) i DM non devono presentare cambiamenti significativi nella progettazione e nella destinazione d’uso;
c) i DM non devono presentare un rischio inaccettabile per la salute o la sicurezza di pazienti, utilizzatori o altre persone o per altri aspetti della protezione della salute pubblica;
d) entro il 26 maggio 2024 il fabbricante deve avere istituito un sistema di gestione della qualità conformemente all’ 10, par. 9 MDR;
e) entro il 26 maggio 2024 il fabbricante o il mandatario deve avere presentato una domanda formale a un ON conformemente all’allegato VII, punto 4.3, primo comma MDR, per la valutazione della conformità del dispositivo (o di un dispositivo destinato a sostituirlo); tale domanda deve quindi contenere tutte le informazioni e dichiarazioni del fabbricante previste dalla pertinente valutazione della conformità di cui agli allegati da IX a XI MDR;
f) entro il 26 settembre 2024 l’ON e il fabbricante devono avere firmato un accordo scritto conformemente all’allegato VII, punto 4.3, secondo comma MDR; tale accordo deve quindi prevedere termini e condizioni chiari e contenere obblighi che consentano all’ON di agire come previsto dal MDR, compreso l’obbligo del fabbricante di informare l’ON delle segnalazioni in materia di vigilanza, il diritto dell’ON di sospendere, limitare o revocare i certificati rilasciati e il dovere dell’ON di adempiere ai propri obblighi di informazione.
Durante il periodo transitorio, gli ON non possono emettere nuovi certificati in base alla Direttiva.
Il fabbricante può, peraltro, dover dimostrare il rispetto delle condizioni previste dall’articolo 120, par. 3 MDR (ad esempio, per accedere al mercato in paesi terzi, o per partecipare a una gara d’appalto). Tale prova può essere fornita tramite:
- un’autodichiarazione che attesti il rispetto delle condizioni per l’estensione, indicando la data di fine del periodo transitorio per il proprio prodotto;
- una “confirmation letter” emessa dall’ON che ha rilasciato il certificato, che attesti la ricezione della domanda di valutazione della conformità (“application”) e la conclusione di un accordo scritto tra le due parti.
La confirmation letter è stata introdotta come strumento per gestire la transazione dei dispositivi legacy verso la piena conformità al MDR; essa dimostra che i DM legacy rispettano determinate condizioni per cui essi possono continuare a essere commercializzati anche se il possesso di valutazione della conformità non è stato ancora completato, e che il loro fabbricante ha intrapreso tutti gli step opportuni per dimostrare la conformità ai requisiti del nuovo contesto normativo. La confirmation letter ha quindi lo scopo di garantire che i pazienti e i professionisti del settore sanitario abbiano accesso a DM medici necessari mentre i fabbricanti completano la transizione alla piena confomità con il MDR.
Nel maggio 2023, l’associazione europea degli ON (Team – NB) ha pubblicato un template di “Notified body confirmation letter“. Esso prevede i seguenti dati essenziali:
- identificativo dell’ON che rilascia la confirmation letter;
- identificativo del produttore, incluso l’SRN number;
- lista dei DM per i quali l’ON che rilascia la lettera è responsabile della sorveglianza appropriata ai sensi della Direttiva;
- lista dei DM per i quali l’ON non è responsabile della sorveglianza appropriata ai sensi della Direttiva.
Come chiarito dalla Commissione nel documento “Q&A on practical aspects related to the implementation of Regulation (EU) 2023/607 amending Regulation (EU) 2017/45” del marzo 2023, ai fini dell’application da presentare entro il 26 maggio 2024, non è necessario un riesame completo della domanda da parte dell’ON prima della firma dell’accordo scritto; ad es. non è necessario che la domanda includa la documentazione tecnica di ciascun DM oggetto della stessa.
Pertanto, la domanda di richiesta di valutazione della conformità a un ON deve includere soltanto le informazioni utili ad identificare chiaramente il fabbricante e i DM e quelle strettamente necessarie all’ON per concludere l’accordo, ovvero:
- le informazioni utili ad identificare il fabbricante e i dispositivi da valutare, eventualmente anche quelli che andranno a sostituire un dispositivo legacy;
- le informazioni necessarie all’ON per finalizzare l’accordo;
- gli elementi elencati nella valutazione di conformità di cui agli allegati da IX a XI MDR;
- le informazioni che consentano all’ON di verificare la qualificazione dei prodotti come dispositivi, la loro rispettiva classificazione e la procedura di valutazione della conformità prescelta;
- una schedulazione delle tempistiche, in accordo con l’ON, per l’eventuale fornitura della documentazione tecnica non fornita al momento della presentazione della domanda, e di qualsiasi altra informazione pertinente;
- la documentazione sul Sistema di Gestione della Qualità (QMS) del fabbricante.
Fino al 26 settembre 2024, l’Organismo notificato secondo la Direttiva rimane responsabile della sorveglianza dei DM che ha certificato. A partire da tale data, l’ON secondo MDR, designato dal fabbricante, assume la responsabilità della sorveglianza. E’ quindi necessario un accordo tripartito tra fabbricante, Organismo secondo la Direttiva e ON secondo l’MDR, per stabilire le modalità di trasferimento della sorveglianza.
La Commissione ha precisato che tale accordo deve rispettare i principi di cui all’art. 58, par. 1 MDR, disciplinare anche il trasferimento della documentazione tra i due Organismi, e contemplare la possibilità per l’ON di sospendere o ritirare un certificato rilasciato dall’Organismo precedente, se debitamente giustificato.
Sull’etichettatura, compresa la marcatura CE, può continuare ad essere indicato il numero dell’Organismo che ha rilasciato il certificato ai sensi della Direttiva e che è ancora valido. Il fabbricante può comunque modificare l’etichettatura dei dispositivi preesistenti indicando il numero dell’ON a cui è stata presentata domanda formale ai sensi dell’MDR.
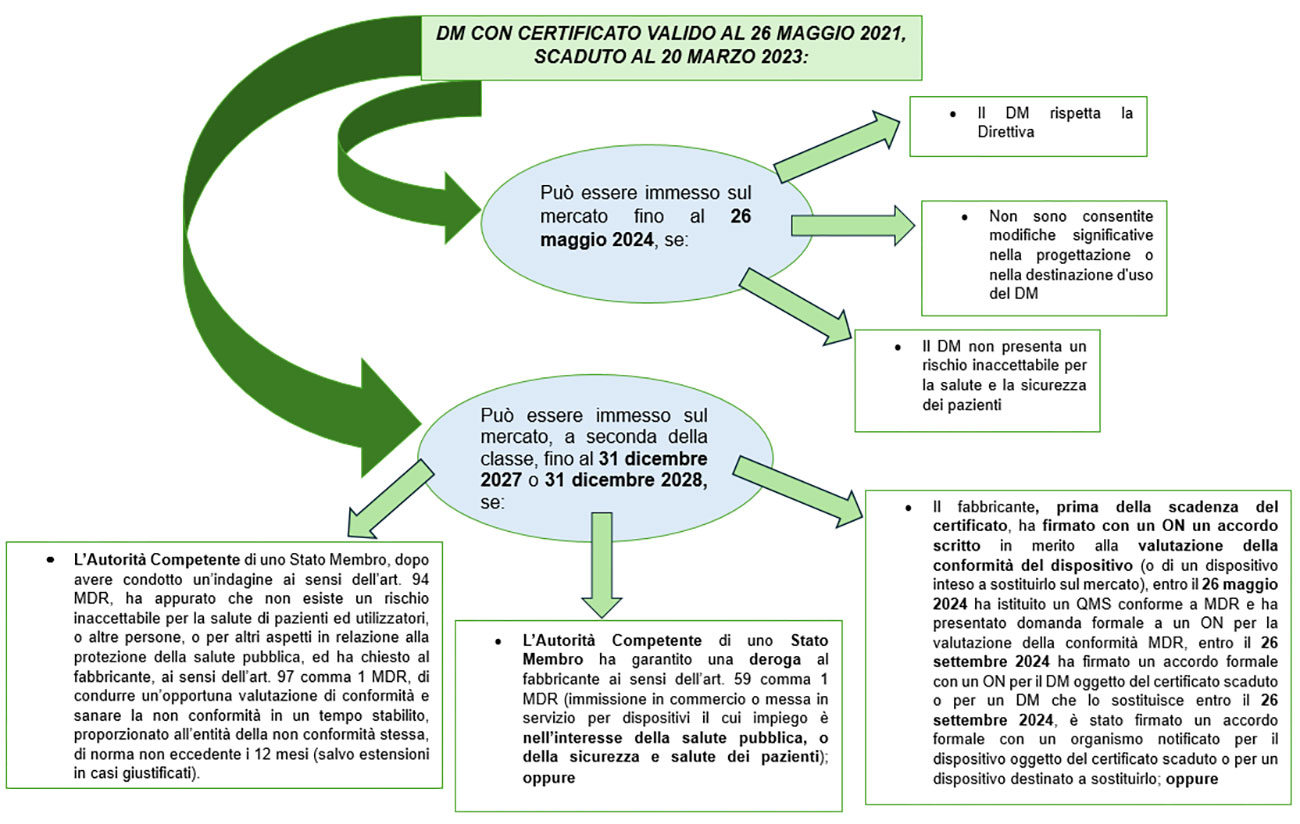
Se invece un DM, alla data del 26 maggio 2021, aveva un certificato CE valido secondo la Direttiva, ma scaduto prima della data di pubblicazione del Regolamento (UE) 2023/607 (20 marzo 2023), l’immissione in commercio di tale certificato è comunque possibile fino al 31 dicembre 2027 (per le classi III e IIb impiantabili) e fino al 31 dicembre 2028 (per le classi Iib non impiantabili, Iia, I sterile o con funzione di misura), ma solo se:
- il fabbricante, prima della scadenza del certificato, ha firmato con un ON un accordo scritto in merito alla valutazione della conformità del dispositivo (o di un dispositivo inteso a sostituirlo sul mercato); in tale ipotesi, può essere emessa una Confirmation letter da parte dell’ON, la quale attesti che l’accordo scritto ai sensi del MDR è stato firmato prima della scadenza del certificato e per dimostrare dunque che anche i dispositivi con certificato scaduto possono continuare a beneficiare dell’estensione del periodo di transizione; oppure
- l’Autorità Competente di uno Stato Membro ha garantito una deroga al fabbricante ai sensi dell’art. 59 comma 1 MDR (immissione in commercio o messa in servizio per dispositivi il cui impiego è nell’interesse della salute pubblica, o della sicurezza e salute dei pazienti); oppure
- l’Autorità Competente di uno Stato Membro, dopo avere condotto un’indagine ai sensi dell’art. 94 MDR, ha appurato che non esiste un rischio inaccettabile per la salute di pazienti ed utilizzatori, o altre persone, o per altri aspetti in relazione alla protezione della salute pubblica, ed ha chiesto al fabbricante, ai sensi dell’art. 97 comma 1 MDR, di condurre un’opportuna valutazione di conformità e sanare la non conformità in un tempo stabilito, proporzionato all’entità della non conformità stessa, di norma non eccedente i 12 mesi (salvo estensioni in casi giustificati).
In ogni caso, i fabbricanti di dispositivi legacy sono tenuti al rispetto degli obblighi di sorveglianza post-market e sorveglianza del mercato da parte dell’ON, vigilanza e di registrazione degli operatori economici previsti dal MDR. Il fabbricante deve quindi, entro il 26 maggio 2024:
- istituire un sistema di gestione della qualità conforme alle prescrizioni dell’art. 10 MDR;
- presentare una domanda formale a un ON;
- sottoscrivere un contratto per la valutazione della conformità del DM (o di un DM destinato a sostituirlo) entro il 26 settembre 2024.
Ai sensi dell’art. 120 MDR, l’ON non è responsabile delle attività di valutazione della conformità svolte dall’Organismo che ha rilasciato il certificato secondo la Direttiva; le sue attività sono limitate all’esecuzione della sorveglianza appropriata di cui all’art. 120 par. 3 sexies MDR. L’Organismo che ha rilasciato il certificato secondo la Direttiva continua quindi ad essere responsabile dell’appropriata sorveglianza rispetto ai requisiti applicabili relativi ai DM che ha certificato; tuttavia, anche prima del 26 settembre 2024 il fabbricante può concordare che l’ON diventi già responsabile della sorveglianza del DM legacy. In ogni caso, da tale data l’ON che ha firmato l’accordo con il fabbricante diventerà responsabile della sorveglianza dei corrispondenti dispositivi legacy (pur non essendo stati certificati da questi), fino alla data del periodo di transizione.
2.3.1 Quali modifiche possono essere apportate ai dispositivi legacy
Come si è accennato, un dispositivo legacy coperto da un certificato emesso nel vigore della Direttiva può essere immesso sul mercato beneficiando del periodo transitorio qualora non siano intervenuti cambiamenti significativi nella progettazione e nella destinazione d’uso del DM, e non si sia verificato un rischio inaccettabile per la salute o sicurezza.
Secondo la Guida del Medical Device Coordination Group (MDCG) 2020-3, la rilevanza giuridica dei cambiamenti sui DM ai fini dell’art. 120 MDR si ha in presenza di due requisiti:
- il cambiamento deve essere inerente alla destinazione d’uso e/o al design;
- tale cambiamento deve essere “significativo”.
Non sono considerate significative le modifiche amministrative (modifiche del nome, dell’indirizzo o della forma giuridica del fabbricante o modifiche del rappresentante autorizzato), il processo generale di realizzazione dei prodotti e il trasferimento o l’aggiunta di nuovi siti di produzione o alcune modifiche al sistema di gestione della qualità, purché siano mantenute le condizioni per le quali è stata rilasciata la certificazione di valutazione della conformità.
Tuttavia, un cambiamento è considerato “significativo” se un fabbricante trasferisce semplicemente DM certificati ai sensi della Direttiva a un altro fabbricante che li commercializza con il proprio nome (c.d. VR o OBL).
I cambiamenti alla destinazione d’uso del prodotto non sono considerati significativi quando comportano la restrizione o eliminazione di alcune indicazioni d’uso o la restrizione o eliminazione di alcuni ambiti di applicazione (come siti anatomici o modo d’impiego), significativi quando rappresentano un’estensione della destinazione d’uso o contengono l’aggiunta di alcune indicazioni d’uso, possibili utilizzatori e/o possibili applicazioni cliniche del prodotto.
I cambiamenti apportati al design del dispositivo non si considerano significativi quando non riguardano il meccanismo di controllo integrato nel prodotto, il suo principio di funzionamento, la fonte di energia o il sistema di allarme. Non devono essere alterate sicurezza, performance o usabilità, e i cambiamenti non devono incidere negativamente sul rapporto rischi/benefici del dispositivo.
Per quanto riguarda le specifiche del prodotto e l’etichettatura, la linea guida riporta alcuni esempi di cambiamenti considerati non significativi:
- cambio di colore del dispositivo;
- cambiamenti che rientrano nel range di caratteristiche già certificate;
- modifiche all’etichetta per migliorare la comprensione di alcuni avvertimenti o informazioni critiche da parte dell’utilizzatore finale.
- modifiche all’imballaggio esterno.
Per quanto concerne un componente del dispositivo, non sono considerati significativi la modifica di un componente elettronico a causa, ad esempio, di obsolescenza, o il cambio di dimensione o forma geometrica di un pulsante di allarme. Per quanto riguarda la fonte di energia, non è considerato significativo il cambio di tipo di batterie o la modifica di una batteria in un sistema ricaricabile.
Sono invece considerati significativi:
- la modifica di un dispositivo da manuale a gestito da un software.
- la modifica delle funzioni di misura del dispositivo.
- l’eliminazione o l’aggiunta di un sistema di allarme.
2.4 Gli obblighi dei distributori e importatori di dispositivi legacy nel periodo transitorio
Durante il periodo transitorio, gli importatori e distributori che commercializzano DM legacy sono soggetti alle previsioni di cui all’art. 120 MDR (ovvero alla sorveglianza, vigilanza e registrazione).
Inoltre, come precisato dalla Mdcg 2021-25, tali operatori devono porre in essere i seguenti adempimenti:
- verifica della conformità del dispositivo alle prescrizioni della Direttiva, ovvero che il DM sia corredato di idonea e conforme documentazione tecnica, sia stata correttamente redatta la sua dichiarazione di conformità (secondo la Direttiva) e, qualora di classe superiore alla I, sia corredato di certificato CE regolarmente emesso dall’Organismo secondo la Direttiva, ancora valido; qualora rilevino la non conformità del dispositivo, importatori e distributori devono astenersi dal commercializzarlo fino a quando le irregolarità non vengano sanate;
- comunicazione all’Autorità competente quando ritengano(secondo la propria esperienza professionale) o abbiano motivo di credere (alla luce delle informazioni acquisite) che un DM sia idoneo a produrre un rischio grave per la salute (ovvero sia probabile il verificarsi di un danno di entità considerevole) e/o comunque possa risultare falsificato;
- registrazione su Eudamed e verifica di registrazione del DM da parte dell’importatore (tale prescrizione non è ancora vincolante: ad oggi è possibile registrarsi volontariamente soltanto ai primi due moduli di Eudamed dedicati alla registrazione degli operatori economici e dei codici UDI);
- tenuta di un registro dei reclami, dei dispositivi non conformi, dei richiami e dei ritiri effettuati e comunicazione delle informazioni utili agli altri operatori economici per l’esame dei reclami ricevuti;
- comunicazioni al fabbricante e al mandatario relative ad un dispositivo non conforme e alle Autorità competenti se i DM presentano un rischio grave e, nel caso, anche all’ON; quando il DM presenti un rischio grave, importatore e distributore devono anche dare comunicazione alle Autorità competenti fornendo informazioni precise sulla non conformità e sulle eventuali azioni correttive intraprese (ad es. il blocco della commercializzazione attraverso provvedimenti di richiamo, ritiro e interruzione del dispositivo);
- cooperazione con gli altri operatori economici e l’Autorità competente per l’adozione di azioni correttive necessarie;
- trasmissione al fabbricante e al mandatario di eventuali segnalazioni e reclami ricevuti, a seguito del verificarsi di un “incidente” (ovvero, ai sensi dell’art. 2 lett. 64 MDR, di “qualsiasi malfunzionamento o alterazione delle caratteristiche o delle prestazioni di un dispositivo messo a disposizione sul mercato, compreso l’errore d’uso determinato dalle caratteristiche ergonomiche, come pure qualsiasi inadeguatezza nelle informazioni fornite dal fabbricante e qualsiasi effetto collaterale indesiderato”);
- cooperazione con l’Autorità competente per attenuare/ridurre i rischi del dispositivo, anche se ciò dovesse impedirne la commercializzazione; in questi casi l’Autorità competente potrà definire con importatore e/o distributore tutte le più opportune misure correttive.
3. L’ambito di applicazione del MDR
3.1 La definizione di dispositivo medico
L’MDR si applica anzitutto ai DM e i relativi accessori, nonché ai dispositivi destinati alle indagini cliniche.
I DM sono definiti come “qualunque strumento, apparecchio, apparecchiatura, software, impianto, reagente, materiale o altro articolo, destinato dal fabbricante a essere impiegato sull’uomo, da solo o in combinazione, per una o più delle seguenti destinazioni d’uso mediche specifiche:
- diagnosi, prevenzione, monitoraggio, previsione, prognosi, trattamento o attenuazione di malattie;
- diagnosi, monitoraggio, trattamento, attenuazione o compensazione di una lesione o di una disabilità;
- studio, sostituzione o modifica dell’anatomia oppure di un processo o stato fisiologico o patologico;
- fornire informazioni attraverso l’esame in vitro di campioni provenienti dal corpo umano, inclusi sangue e tessuti donati, e che non esercita nel o sul corpo umano l’azione principale cui è destinato mediante mezzi farmacologici, immunologici o metabolici, ma la cui funzione può essere coadiuvata da tali mezzi.
Rientrano altresì nella definizione di DM anche i seguenti prodotti:
- dispositivi per il controllo del concepimento o il supporto al concepimento,
- prodotti specificamente destinati alla pulizia, disinfezione o sterilizzazione dei dispositivi.
L’MDR non si applica invece ai dispositivi medico-diagnostici in vitro, disciplinati dal Regolamento UE n. 2017/746 entrato in vigore il 26 maggio 2022.
L’MDR non si applica altresì:
- ai medicinali;
- ai dispositivi che contengono o sono costituiti da sostanze biologiche vitali;
- ai dispositivi che contengono emoderivati, plasma o cellule ematiche e gli alimenti.
La distinzione tra DM (soggetti alla disciplina dell’MDR) e medicinali (soggetti alla disciplina della Direttiva 2001/83/CE, attuata dal D.lgs. 2019/2006 , c.d. Codice del Farmaco) può tuttavia essere problematica, data l’esistenza dei DM a base di sostanze (medicinali); queste ultime consistono, come chiarito dalle linee guida MDCG 2022-5, in ogni materia indipendentemente dall’origine umana (come il sangue umano e suoi derivati), animale (come microrganismi, animali interi, parti di organi, secrezioni animali, tossine, sostanze ottenute per estrazione, prodotti derivati dal sangue), vegetale (come microrganismi, piante, parti di piante, secrezioni vegetali, sostanze ottenute per estrazione) o chimica (come elementi, materie chimiche naturali).
Secondo le linee guida, una sostanza è un medicinale quando presenta un meccanismo d’azione farmacologico, immunologico o metabolico, indipendentemente dalla destinazione d’uso stabilita (proposta) dal fabbricante, dalla quantità della sostanza, metodo e via di somministrazione.
Se dunque il fabbricante dimostra che la sostanza non presenta un meccanismo d’azione farmacologico, immunologico o metabolico, il prodotto non può essere qualificato come medicinale e rientra nella nozione di DM. Se invece si accerti l’esistenza di un meccanismo d’azione farmacologico, immunologico e/o metabolico, occorre verificare se il meccanismo d’azione della sostanza assolva funzione principale o accessoria rispetto all’azione esercitata dal (componente) DM; nel primo caso il prodotto è un medicinale (soggetto alla relativa normativa), nel secondo caso (quando cioè l’azione prodotta nel/sul corpo umano o sui suoi componenti si limita a supportare la destinazione d’uso del componente DM) il prodotto è un DM, soggetto alla disciplina speciale prevista dall’art. 52, par. 9, MDR.
Per determinare in concreto se la sostanza assolva una funzione accessoria, le linee guida richiedono al fabbricante di eseguire un’attenta valutazione tecnico-scientifica basata su dati oggettivi e compatibili con lo stato dell’arte, considerando in particolare la capacità della sostanza e la quantità disponibile per il corpo umano e/o suoi componenti.
Le conseguenze di una errata qualificazione del prodotto sono rilevanti.
Se infatti il fabbricante applica la procedura speciale prevista dal dall’art. 52, par. 9, MDR ad un prodotto che doveva essere qualificato come medicinale, lo stesso potrebbe essere soggetto ad una duplice sanzione pecuniaria: la sanzione prevista per la violazione del MDR e la sanzione prevista dell’art. 148, comma 1, del Codice del Farmaco per aver commercializzato un prodotto medicinale senza la necessaria autorizzazione (c.d. AIC) (da 3 mila a 18 mila euro).
Viceversa, qualora il fabbricante applichi erroneamente il codice del Farmaco ad un prodotto che doveva essere qualificato come DM, allo stesso potrebbe essere irrogata la sanzione pecuniaria prevista dall’art. 27, comma 32, D.lgs. 137/2022 e/o andare incontro a richieste di risarcimento danni per concorrenza sleale ai sensi dell’art. 2598 c.c. da parte di aziende concorrenti.
In base alla definizione contenuta nel MDR, la qualifica di un dispositivo come DM dipende soprattutto dalla sua destinazione d’uso; per tale motivo, come esplicitamente indicato nell’art. 6 MDR, anche i dispositivi venduti online e quelli utilizzati per fornire un servizio diagnostico o terapeutico tramite i servizi della società dell’informazione sono disciplinati dal MDR.
Per lo stesso motivo, anche i dispositivi fabbricati e utilizzati esclusivamente in istituzioni sanitarie sono qualificati come DM, se hanno una o più destinazioni d’uso indicate nella definizione. Ciononostante, per tali dispositivi non è necessario applicare tutte le prescrizioni del MDR se sono soddisfatte le condizioni indicate nell’art. 5, paragrafo 5 (v. par.4).
3.2. I dispositivi medici non aventi destinazione d’uso medica
L’MDR si applica altresì ad alcuni prodotti che non hanno una destinazione d’uso medica, i quali, pur non rientrando nella nozione di dispositivo medico, possono presentare analoghi rischi per i consumatori.
Tali prodotti sono elencati nell’allegato XVI e comprendono:
- lenti a contatto o altri elementi destinati a essere introdotti nel o sull’occhio (ad esempio lenti colorate);
- prodotti destinati a essere introdotti anche solo parzialmente nel corpo mediante strumenti invasivi di tipo chirurgico per modificare l’anatomia o per la fissazione di parti del corpo (esclusi prodotti per tatuaggi e piercing) come ad esempio protesi mammarie per estetica;
- sostanze utilizzate per filling facciali o altri filling cutanei o per le mucose attraverso iniezione sottocutanea, sottomucosa o intradermica (esclusi quelli per i tatuaggi) come ad esempio il filler dermico;
- apparecchiature per ridurre, rimuovere o distruggere il tessuto adiposo (ad esempio macchine per la liposuzione, lipolisi o l’ipoplastica);
- apparecchiature che emettono radiazioni elettromagnetiche ad alta intensità (ad es. infrarossi, luce visibile e ultravioletti) destinate a essere utilizzate sul corpo umano, comprese fonti coerenti e non coerenti, monocromatiche e ad ampio spettro (ad esempio laser e apparecchiature a luce pulsata ad alta intensità per foto ringiovanimento cutaneo, tatuaggio o epilazione o altro trattamento dermico);
- attrezzature per la stimolazione cerebrale che applicano correnti elettriche o campi magnetici o elettromagnetici che attraversano il cranio per modificare l’attività neuronale del cervello come, ad esempio, apparecchiature per la stimolazione magnetica transcranica (non invasive chirurgicamente).
Rientrano quindi nella definizione di DM ai sensi del MDR anche dispositivi aventi utilizzo cosmetico/estetico, come ad esempio le lenti a contatto di tipo cosmetico, le protesi di tipo estetico, i filler utilizzati per usi estetici, gli apparecchi laser per la rimozione dei tatuaggi, etc.
In vigenza della Direttiva, i prodotti di cui sopra erano per lo più di libera vendita; oggi devono rispettare, in generale, le prescrizioni del MDR, con alcune differenze. La realizzazione dei prodotti dell’Allegato XVI deve infatti rispettare le “Specifiche Comuni” (SC) stabilite appositamente per questi prodotti dal Regolamento di Esecuzione UE 2022/2346 del 1° dicembre 2022 (allegati dal I al VII), e non quelle emanate per gli altri DM (art. 9 par. 4 MDR).
Come evidenziato dalla Circolare del Ministero della Salute del 26 novembre 2025, per i prodotti dell’Allegato XVI per i quali non sono state definite specifiche comuni da parte della Commissione, l’MDR non si applica. Sul punto la MDCG 2023-5 (Guidance on qualification and classification of Annex XVI products) chiarisce che i prodotti esclusi dalle SC spesso non soddisfano i criteri per essere considerati dispositivi dell’Allegato XVI.
Per quanto riguarda le regole di classificazione, trova applicazione l’Allegato VIII dell’MDR. Tuttavia, per i prodotti attivi di cui ai punti 4, 5 e 6 dell’Allegato XVI (apparecchiature per ridurre, rimuovere o distruggere il tessuto adiposo; apparecchiature che emettono radiazioni elettromagnetiche ad alta intensità, attrezzature destinate alla stimolazione cerebrale) il Regolamento di esecuzione (UE) 2022/2347 ha introdotto regole di classificazione specifiche, che innalzano il livello di sorveglianza e controllo. In particolare:
- le apparecchiature destinate a essere utilizzate per ridurre, rimuovere o distruggere il tessuto adiposo sono riclassificate nella Classe IIb.
- le attrezzature destinate alla stimolazione cerebrale sono riclassificate nella Classe III.
- le apparecchiature che emettono radiazioni elettromagnetiche ad alta intensità destinate a essere utilizzate sul corpo umano per trattamento dermico sono riclassificate nella Classe IIb, a meno che non siano destinate unicamente all’epilazione, nel qual caso sono riclassificate nella Classe IIa.
Ai sensi dell’art. 1 par. 2 MDR, i prodotti dell’Allegato XVI devono rispettare l’MDR a partire dalla data di applicazione delle relative SC; ai sensi quindi dell’art. 3 del Regolamento 2022/2346 le SC sono applicabili dal 22 giugno 2023. Tuttavia, analogamente all’articolo 120 MDR, anche per i prodotti dell’Allegato XVI il Regolamento 2022/2346 ha previsto un periodo transitorio, poi modificato dal Regolamento di Esecuzione (UE) 2023/1194 del 20 giugno 2023, per i prodotti immessi sul mercato prima del 22 giugno 2023. In particolare:
- per i prodotti già commercializzati che continuano a essere conformi alle normative previgenti e per i quali non vi siano cambiamenti significativi nella progettazione o destinazione d’uso, si applicano due distinti periodi di transizione a seconda degli obblighi di conformità: a) scadenza 31 dicembre 2029 per i prodotti che richiedono una indagine clinica, a condizione che entro il 22 giugno 2024 lo sponsor abbia ricevuto la conferma che la domanda per l’indagine clinica sia completa, che l’indagine clinica sia avviata entro il 23 dicembre 2024 e che il fabbricante e l’ON sottoscrivano un accordo scritto per la valutazione della conformità entro il 1° gennaio 2028; b) scadenza 31 dicembre 2028 per i prodotti che non richiedono indagine clinica, se il fabbricante e l’ON sottoscrivano un accordo scritto per la procedura di valutazione della conformità entro il 1° gennaio 2027;
- per prodotti già certificati ai Sensi della Direttiva, il periodo di transizione termina il 31 dicembre 2027 (per Classe III o impiantabili di Classe IIb) o il 31 dicembre 2028 (negli altri casi), in base alla classe di rischio del DM; come per gli altri DM, il fabbricante, per beneficiare di tali termini, deve rispettare le condizioni previste dall’art. 2 par. 3 del Regolamento 2022/2346, che richiama l’art. 120 par. 3 dell’MDR, (come modificato dal Regolamento (UE) 2023/607).
Per quanto concerne il Sistema di Gestione della Qualità (SGQ), entro il 26 maggio 2024 il fabbricante deve aver istituito un SGQ (ai sensi dell’art. 10 par. 9 MDR) e aver presentato una domanda formale all’ON per la valutazione della conformità, ed entro il 26 settembre 2024 il fabbricante e l’ON devono aver hanno sottoscritto un accordo scritto.
Il Regolamento (UE) 2022/2346 non indica come debbano essere svolte le attività di sorveglianza del mercato da parte delle Autorità competenti durante i periodi transitori per i prodotti senza una destinazione d’uso medica. La Circolare del Ministero della Salute del 26 novembre 2025 ha chiarito in proposito che tale attività resta disciplinata dalla normativa tecnica armonizzata applicabile, a seconda delle specifiche del singolo prodotto considerato, da valutare caso per caso.
La Circolare chiarisce altresì che le competenze in materia di sorveglianza del mercato sono esercitate a livello nazionale e, pertanto, il perimetro di attività delle diverse Autorità competenti può variare da uno Stato membro all’altro. In particolare:
- se un prodotto non è immesso sul mercato ai sensi del Regolamento (UE) 2017/745, l’Autorità nazionale responsabile è individuata sulla base delle competenze indicate al D.lgs 157/2022 che adegua la disciplina nazionale al Reg. Ue 2019/1020;
- se un prodotto è immesso sul mercato ai sensi del Regolamento (UE) 2017/745, l’Autorità competente è quella designata per i DM (in Italia il Ministero della salute).
3.3 I dispositivi medici software (SAMD)
Ai sensi dell’art. 2 par. 1 MDR, il software possono essere qualificati come DM (Software As Medical Device – SAMD) quando:
- presentano una destinazione d’uso specifica in area medicale, ai sensi dell’art. 1, lett. a), MDR;
- eseguono elaborazioni complesse dei dati inseriti, generando informazioni mediche nuove e diverse (output) dai dati ricevuti (input);
- vengono utilizzati sull’uomo, fornendo informazioni utili per prendere decisioni a scopo diagnostico o terapeutico (Allegato VIII – Regola 11 del MDR).
Un software può quindi essere definito un SAMD – sia che sia incluso in altro dispositivo medico-hardware (embedded) sia che operi autonomamente (stand-alone) – quando supporta gli operatori sanitari nelle attività di diagnosi e terapia, come nel caso di quelli utilizzati per l’interpretazione, l’analisi e l’elaborazione di dati medici o che producono informazioni riguardanti la misurazione di alcuni parametri utili in ambito diagnostico, destinati ad essere elaborati ed analizzati a scopo medico (ad es. software per il controllo della glicemia).
Non sono invece considerati SAMD i software impiegati, seppur in un contesto legato alla sanità, con uno scopo diverso da quello diagnostico o terapeutico, come ad esempio, le piattaforme destinate alla ricerca semplice o alla sola archiviazione e gestione dei dati del paziente, di dati di monitoraggio o di dati riguardanti le decisioni cliniche o le terapie, come le cartelle cliniche elettroniche, se non impiegate in nessuna attività di diagnosi, prevenzione, controllo, terapia o attenuazione di malattia) o App predisposte a monitorare ed elaborare i parametri fisiologici, l’attività fisica, le calorie bruciate, il battito cardiaco, ma non con uno scopo medico.
Data l’ampia definizione di DM adottata dal MDR, molti più software utilizzati in ambito sanitario sono qualificabili come dispositivi medici rispetto a quanto avveniva in precedenza.
La Guida MDCG 2023-4 “Medical Device Software (MDSW) – Hardware combinations Guidance on MDSW intended to work in combination with hardware or hardware components” ha chiarito gli effetti giuridici derivanti dalla interazione fra software e hardware di supporto o hardware che forniscono i dati, in base ai quali il software riesce a raggiungere la propria destinazione d’uso, come nel caso dei software che elaborano i dati derivanti da dispositivi hardware (ad es. smartphone) o i dispositivi wearable (es. “smartwatch”) i quali raccolgono, ad esempio, il dato di misurazione del battito cardiaco, del livello di ossigenazione del sangue e della temperatura corporea.
Secondo la Guida, qualora un SAMD interagisca con un hardware che è qualificato come DM o come accessorio di DM, il fabbricante del SAMD deve verificare, validare e dimostrare la sicurezza, riproducibilità, compatibilità e interoperabilità del DM o accessorio del DM con cui il SAMD dovrà lavorare in combinazione, incluse le varie configurazioni e varianti. Il fabbricante deve altresì valutare la compatibilità tra il software e l’hardware e implementare dei meccanismi che consentano la comunicazione immediata di eventuali incidenti relativi al DM hardware. Per quanto concerne invece la compliance dell’hardware, il fabbricante del SAMD può fare affidamento sulla conformità dell’hardware rispetto al MDR e pertanto non può essere responsabile per eventuali malfunzionamenti dell’hardware.
Qualora invece il SAMD interagisca con un hardware che non è qualificato come DM o come accessorio di DM, il fabbricante del SAMD ha responsabilità ben maggiori, in quanto, oltre agli obblighi di cui sopra, è anche responsabile per la sicurezza, le prestazioni e la riproducibilità dell’hardware nel suo uso combinato con il SAMD. Pertanto, in tal caso il fabbricante del SAMD può risultare responsabile anche per il malfunzionamento dell’hardware, nonostante non sia stato dallo stesso fabbricato. Inoltre, la documentazione tecnica del SAMD deve identificare e descrivere tutti gli hardware con i quali è compatibile e il risk management dovrà chiaramente indicare anche che impatto l’hardware non dispositivo medico può avere sui requisiti di sicurezza e prestazione del SAMD.
4. DM e Intelligenza Artificiale (AI)
Il Regolamento Ue 2024/1689 sull’Intelligenza Artificiale (“AI Act”) è stato pubblicato sulla Gazzetta Ufficiale europea il 12 luglio 2024, ha aperto una nuova era per i software basati su sistemi di intelligenza artificiale (AI). Il Regolamento prevede una serie di regole per la progettazione, realizzazione e immissione sul mercato dei sistemi di AI, che troveranno applicazione non solo nei confronti dei fornitori stabiliti nella Ue, ma anche nei confronti di quelli extra Ue (articolo 2, lett. a. AI Act).
L’art. 3 dell’AI Act definisce sistema di AI “un sistema automatizzato progettato per funzionare con livelli di autonomia variabili e che può presentare adattabilità dopo la diffusione e che, per obiettivi espliciti o impliciti, deduce dall’input che riceve come generare output quali previsione, contenuti, raccomandazioni o decisioni che possono influenzare ambienti fisici o virtuali”.
Si tratta di una definizione molto ampia, la quale ricomprende qualsiasi software che, per finalità determinate dall’uomo, sia in grado di generare output in grado di influenzare gli ambienti – quindi anche le persone – con cui interagiscono.
Secondo l’Explanatory Memorandum of the updated Oecd definition of an AI system, pubblicato nel marzo 2024 dall’OEDC.AI, un software rientra nella nozione di AI qualora vi siano:
- autonomie variabili (cioè, delle diverse capacità di apprendimento in diverse aree – come computer vision, elaborazione del linguaggio naturale, riconoscimento vocale, sistemi intelligenti di supporto alle decisioni, i sistemi robotici intelligenti);
- capacità di adattamento (cioè, la capacità di continuare ad evolversi attraverso l’interazione diretta (con input e dati) anche dopo l’immissione sul mercato;
- obiettivi espliciti (cioè definiti esplicitamente dall’uomo), impliciti, che possono derivare dalle regole specificate dall’uomo o dai dati di addestramento (in quest’ultimo caso, l’obiettivo finale non è programmato esplicitamente, ma è incorporato attraverso i dati di addestramento e un’architettura di sistemi che impara a emulare quei dati), o non completamente conosciuti in anticipo (sistemi di raccomandazione che usano apprendimento per rinforzo per restringere gradualmente il modello delle preferenze dei singoli utenti);
- capacità di generare output, che possono essere raccomandazioni, previsioni e decisioni.
Se dunque un SAMD presenta le funzionalità di cui sopra, esso rientra nella definizione di “intelligenza artificiale” e deve quindi conformarsi all’AI Act.
L’AI Act distingue tra tre categorie di software di intelligenza artificiale:
- i sistemi di AI vietati (art. 5 AI Act);
- i sistemi di AI considerati ad alto rischio (artt. 6 e ss. AI Act);
- i sistemi di AI per finalità generati (artt. 51 e ss. AI Act).
I SAMD che presentano le funzionalità della AI rientrano per lo più nella categoria dei software ad alto rischio. L’art. 6 AI Act stabilisce che un sistema di AI è considerato ad alto rischio se son o presenti entrambi i seguenti presupposti:
- il sistema di AI è progettato per essere un componente di sicurezza di altro prodotto oppure è esso stesso un prodotto autonomo;
- il prodotto che contiene il componente di sicurezza o il prodotto autonomo sono soggetti a una valutazione della conformità da parte di terzi (gli ON) ai fini dell’immissione sul mercato/messa in servizio ai sensi della normativa di armonizzare dell’Unione elencata nell’allegato I (in cui è indicato anche l’MDR).
Alla luce di quanto sopra, sono soggetti all’AI Act e rientrano nella nozione di AI ad altro rischio i SAMD di Classe IIa, IIb, III (sottoposti alla valutazione di conformità di un ON), mentre i SAMD di Classe I rientrano nei software non ad alto rischio, sempre che non svolgano una delle funzioni elencate nell’Allegato III. In tale Allegato sono elencati anche i sistemi di AI che operano nel settore di “accesso ai servizi privati essenziali e a prestazioni e servizi pubblici essenziali e fruizione degli stessi”, in particolare i sistemi di IA destinati a essere utilizzati:
- dalle autorità pubbliche o per conto di autorità pubbliche per valutare l’ammissibilità delle persone fisiche alle prestazioni e ai servizi di assistenza pubblica essenziali, compresi i servizi di assistenza sanitaria, nonché per concedere, ridurre, revocare o recuperare tali prestazioni e servizi;
- per valutare e classificare le chiamate di emergenza effettuate da persone fisiche o per inviare servizi di emergenza di primo soccorso o per stabilire priorità in merito all’invio di tali servizi, compresi polizia, vigili del fuoco e assistenza medica, nonché per i sistemi di selezione dei pazienti per quanto concerne l’assistenza sanitaria di emergenza.
Per i SAMD classificati ad alto rischio, l’AI Act prevede una serie di obblighi, che entreranno in vigore a partire dal 2 agosto 2027, come previsto dall’articolo 113 dell’AI Act. Tali obblighi riguardano tutti gli operatori del settore, ovvero produttori, distributori, rivenditori e deployer. Con tale ultimo termine, in particolare, l’AI Act (art. 3) designa chiunque (persona fisica o giuridica) utilizzi un sistema di IA nell’ambito di un’attività professionale; definizione, quest’ultima, che può applicarsi sia alle strutture sanitarie (pubbliche e private), sia ai singoli esercenti la professione medica, ove si avvalgano nella propria attività – diagnostica come terapeutica o chirurgica – si SAMD o loro componenti.
A secondo del proprio ruolo, tali soggetti saranno chiamati a conformarsi alle specifiche previsioni alle quali l’AI Act assoggetta la produzione e l’impiego di SAMD ad alto rischio, tra cui, a titolo esemplificativo:
- l’obbligo di adozione di un sistema di gestione del rischio;
- il dovere di garantire elevati standard di qualità dei dati impiegati per addestrare il SAMD;
- obblighi di trasparenza e trasmissione delle informazioni concernenti il SAMD, al fine di limitare i rischi connessi alla sua potenziale opacità;
- il dovere di adottare misure di sorveglianza umana del funzionamento del SAMD, nonché di garantire idonei livelli di accuratezza, robustezza e cybersicurezza.
FAQ – Domande frequenti
Avv. Valerio Pandolfini
Avvocato specializzato in diritto farmaceutico
Sul tema dei Dispositivi Medici abbiamo pubblicato anche:
Abbiamo vasta esperienza nella consulenza e assistenza legale nel campo dei Dispositivi Medici.
Potete visionare QUI alcuni dei più significativi e recenti casi che abbiamo trattato.
Per approfondire i nostri servizi di assistenza e consulenza in tema di Dispositivi Medici, visionate la pagina dedicata del nostro sito.
Le informazioni contenute nel presente articolo hanno carattere generale e non sono da considerarsi un esame esaustivo né intendono esprimere un parere o fornire una consulenza di natura legale. Le considerazioni e opinioni di seguito riportate non prescindono dalla necessità di ottenere pareri specifici con riguardo alle singole fattispecie descritte. Di conseguenza, il presente articolo non costituisce un(né può essere altrimenti interpretato quale) parere legale, né può in alcun modo considerarsi come sostitutivo di una consulenza legale specifica.



